







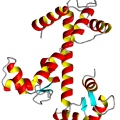
Determining protein folding via Nuclear Magnetic Resonance
In this first focus we explain what is a protein and show one of the techniques to obtain information about their fold, i.e. the spatial distribution of their atoms. At this level we may assume that the proteins are rigid molecules, we will see in a subsequent focus that it is exactly the lack of rigidity that can help proteins to achieve their function, or: Proteins flex to function (Huang and Monteleone, Nature, 2005).
Proteins are macromolecules synthetized by living organisms. They have different functions inside the body: structural, enzimatic, transport or immunitary. They are constituted by a connected senquence of amino acids.

There are twenty amino acids, all having a common backbone formed by a nitrogen followed by two carbide atoms. The type of amino acid depends on the residual, which is the secondary structure connected to the first carbide.
The set of allowable sequences, the genome, is coded inside the DNA. The first decoding of the human DNA dates back to the beginning of this century, it has now become a routine process.
Proteins may be thought as rigid bodies as a first approximation. Their shape, the fold, is the result of some energy minimum, with a merit function including nuclear and electromagnetic forces. Since there might be thousands of atoms in a protein, the effective search for the minimum is not an easy process. We might keep fixed the variables which need a high energy to change, such as the distance between chemically bounds atoms. If we only consider the backbone, we may reduce to two degrees of freedom per amino acid. A protein with 200 amino acids would then have 400 degrees of freedom, which is anyway a large number.
In order to achieve better results we may measure some quantities about the protein, and use the measurements as constraints for the fold. Different types of measurements may be obatined, depending also if the protein is crystallized or in solution. In the second case, which is closer to physiological conditions, we may use the NMR (Nuclear Magnetic Resonance) as a tool to obtain different measurements. Most common are the NOE (Nuclear Overhauser Effect), whch are distances between hydrogen atoms of the protein.
We may often have a paramagnetic metal ion as part of the protein. This may happen because the atom is already part of the protein, as a result of the substitution of a diamagnetic atom, or via a tag, a small rigid molecule containing the paramagnetic atom which attach to the protein molecule always in the same place. In these cases, we may measure the RDC (Residual Dipolar Coupling) and the PCS (Pseudo Contact Shift), depending respectively on the orientation of selected dipoles (typically the N-H or C-H couples) and on the vector joining the metal and selected hydrogen atoms.
Through these measurements it is possible to determine the spatial placement of some parts of the backbone which, because of their sequence, assume a characteristic fixed shape, such as the alpha-helices.
Determining the fold of a protein is a first necessary step to study the oscillations of the fold, i.e. the flexibility of these molecules. The flexibility is a key feature in order to understand the functioning of these molecules in physiological conditions. The determination of the flexibility is the subject of this second focus.



